Congenital adrenal hyperplasia - Clinical features and management
This guide will help you with
- Understanding what is CAH?
- What is the pathophysiology?
- What causes adrenal hypertrophy?
- What is the enzyme pathway for steroids?
- How to investigate CAH?
- Why renin is increased in CAH?
- A quick review of management
Frequently asked question
What are the Clinical features and management of a child with congenital adrenal hyperplasia?
What is Congenital adrenal hyperplasia (CAH)?
Congenital adrenal hyperplasia (CAH) is a family disorders of cortisol biosynthesis. Cortisol deficiency increases the secretion of corticotropin (ACTH), which in turn leads to adrenocortical hyperplasia and overproduction of intermediate metabolites.
Genetics
Congenital adrenal hyperplasia (CAH) is a inherited as autosomal recessive disorders which means one affected gene is needed from each parent to cause the disease (25% probability)
Pathophysiology
But before we head onto that, let us understand...
What do adrenal glands do?
They take up cholesterol and convert it into various end products using various enzymes in the various zone ( Zona glomerulosa, fasciculata and reticularis)
P.S. This chart is always a headache!

What happens in CAH
Depending on the enzymatic step that is deficient, there may be signs, symptoms, and laboratory findings associated with deficiency of end product and excess of pre-products (substrate which was going to be converted into end products, now accumulates as there is no enzyme for conversion).
These deficient end products and excess pre-product may cause
- Mineralocorticoid deficiency or excess
- Incomplete virilization or premature puberty in affected males and
- Virilization or sexual infantilism in affected females.
But wait, why does the adrenal hypertrophy then?
Because of HPA axis.
This is a Normal HPA axis
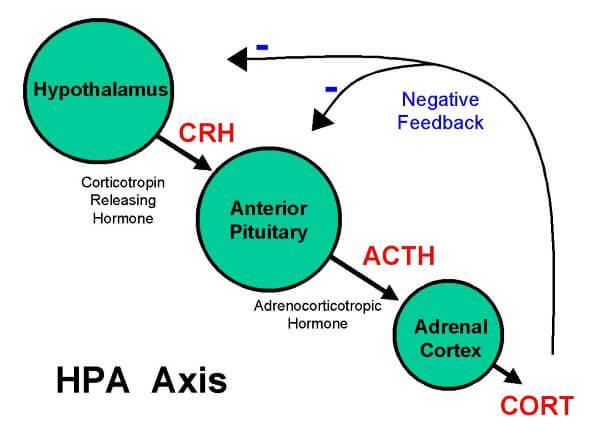
|
| HPA axis, Source: Wikimedia commons |
Low cortisol levels stimulate the hypothalamus which stimulates the pituitary to release ACTH.
Continued ACTH release stimulates the adrenal gland excessively which releases more and more androgen and also gets hypertrophied.
More than 90% of congenital adrenal hyperplasia (CAH) cases are caused by 21-hydroxylase deficiency.
Honestly, there are only two types you need to know for the exam
- 21 hydroxylase 21 0HD – 90-95% of cases
- 11 beta Hydroxylase (11ß-OHD) – 5-8% of cases
| Enzyme Deficiency | 21 hydroxylase | 11B hydroxylase | 17 a hydroxylase |
|---|---|---|---|
| Aldosterone | ↓ | ↓ | ↑ |
| Androgen | ↑ | ↑ | ↓ |
| Potassium | ↑ | ↓ | ↓ |
| Blood pressure | ↓ | ↑ | ↑ |
Clinical features of congenital adrenal hyperplasia (CAH)
CAH cause a wide range of symptoms ranging from asymptomatic disease to life-threatening shock depending upon the severity of enzyme deficiency.
The clinical features are due to ( refer to pathophysiology)
- Low cortisol (End product deficiency)
- Excess androgen (excess accumulation of substrate)
- High ACTH (HPA axis stimulation)
1) Cortisol and aldosterone deficiency
Typical symptoms of Cortisol and aldosterone deficiency are
- Progressive weight loss,
- Anorexia,
- Vomiting,
- Dehydration,
- Weakness,
- Hypotension,
- Hypoglycemia,
- Hyponatremia, and
- Hyperkalemia.
The salt-wasting variant needs to be picked up early to prevent a salt-losing adrenal crisis. These problems typically first develop in affected infants at approximately 10-14 days of age. Without treatment, shock, cardiac arrhythmias, and death may occur within days or weeks.
2) Androgen excess
A. Prenatal androgen excess
This problem begins in affected fetuses by 8-10 wk of gestation and leads to abnormal genital development in females. This manifests as
- Labial fusion,
- Clitoromegaly,
- Urogenital sinus,
- Sexually dimorphic behavior (Masculine play with decreased interest in maternal roles).
B. Postnatal androgen excess
This manifests as
- Rapid somatic growth and accelerated skeletal maturation (tall in childhood with stunted adult stature).
- Excess muscular development, pubic and axillary hair, enlarged penis with relatively small testes).
4) Excess ACTH
Along with adrenal hypertrophy, excessive ACTH also stimulates melanocytes and causes hyperpigmentation. Scrotal hyperpigmentation maybe your first clue in diagnosing CAH.
5) Adrenomedullary dysfunction
Blunted epinephrine responses, decreased blood glucose, and lower heart rates with exercise are the main signs.
21 Hydroxylase deficiency
It is the most common type
- Complete enzyme deficiency - Early-onset - excess androgen - virilization of a baby girl, precocious puberty in boys
- Partial deficiency - Late-onset - Menstrual irregularity and hirsutism in young girls
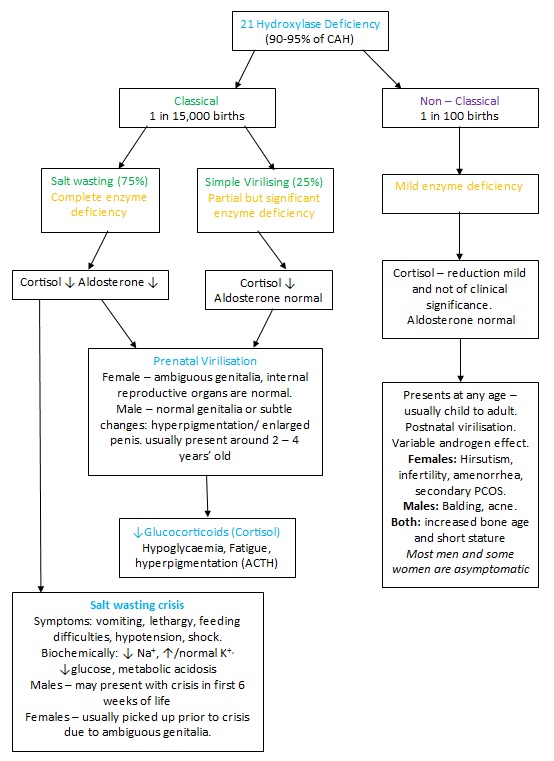
|
| 21 hydroxylase deficiency. Source: Unknown |
Clinical Investigations in congenital adrenal hyperplasia
Electrolytes
- Serum electrolytes - hyponatremia, hyperkalemia, metabolic acidosis.
- Blood sugar – Hypoglycemia
Hormonal assays
- Elevated 17 (OH) progesterone - If 21 hydroxylase is deficient 17 OHP will accumulate (Substrate)
- Low serum cortisol - (end product)
- Elevated renin - Low serum aldosterone activate the renin-angiotensin-aldosterone axis (RAAS)
Why renin is elevated in CAH?
Low serum aldosterone activates renin-angiotensin-aldosterone axis (RAAS)
Genital anatomy
USG abdomen and pelvis to look for internal genital organs ( Ovaries and uterus) in phenotypic females.
Genetic testing
Karyotyping to determine the genetic sex of the child and also to detect any chromosomal abnormality.
Prenatal tests and newborn screening
- Prenatal diagnosis – DNA analysis for the CYP21 gene can be done for diagnosing CAH antenatally.
- 17 (OH) progesterone levels in heel prick dried blood samples is a commonly done screening test to rule out the most common variety of CAH.
Differential Diagnosis
- Pyloric stenosis - CAH may present as a “simple” vomiting and dehydration.
- Androgen insensitivity syndrome,
- Hyperprolactinemia,
- Cushing syndrome, and
- Adrenal tumor
Management in CAH
The principle of treatment is
- Replace the deficient steroids (cortisol and/or aldosterone) and
- Reduce excess androgens.
- Stop HPA axis overstimulation of adrenals by replacing cortisol and reducee the amount of androgen produced.
Treat Shock if present
The priority in the severe salt-wasting variety is shock management. Follow general principles of shock management with emphasis on
- Hyponatremia
- Hypoglycemia
- Hyperkalemia
- Cortisol in stress dose. Hydrocortisone
1) Glucocorticoid replacement
Typically 15-20 mg/m2/day of hydrocortisone is administered via oral route in 3 divided doses. This suppresses excessive production of androgens by the adrenal cortex and thus minimizes problems such as excessive growth and skeletal maturation and virilization.
Stress dose required during periods of stress e.g, infection, surgery.
Regular growth monitoring and pubertal development assessment are required to adjust the dose.
2) Mineralocorticoid replacement
Mineralocorticoid replacement is required in salt-wasting disease. High doses of fludrocortisone, usually 0.1-0.3 mg daily in 2 divided doses but occasionally up to 0.4 mg daily are required. Often sodium supplementation is also provided.
Serum electrolytes need to be monitored initially daily after beginning the therapy.
3) Surgical Management of Ambiguous Genitals
- Significantly virilized females usually undergo surgery between 2-6 mo of age.
- If there is severe clitoromegaly, the clitoris is reduced in size, with partial excision of the corporal bodies and preservation of the neurovascular bundle.
- Vaginoplasty and correction of the urogenital sinus usually are performed at the time of clitoral surgery.
- Revision surgery is often necessary for adolescence.
4) Prenatal treatment
- Dexamethasone 20mcg/Kg of pre-pregnancy weight is started at 6 weeks of gestation for at-risk pregnancies.
- A Chorionic villus biopsy is then done at 11-12 weeks to confirm the sex of the fetus.
- If the sex of the fetus is female, Dexamethasone is continued till term.
Follow up
- Height-weight, growth monitoring
- Monitoring blood pressure
- Pubertal development
- Behavioral and psychosocial

In mood of reading more Q and A?
- Burns - short question
- Cold injuries in children
- Carbon monoxide poisoning
- Acute Otitis media - short question
- ARDS - Long question
- Congenital cataract - short question
Author

Shailesh Gophane | DCH DNB Pediatrics
Shailesh completed his Pediatric residency from Port Trust Hospital Mumbai after completing DCH from J.J. Hospital, Mumbai
1 comment
🩺 Help us refine this article — share corrections or additional information below. Let's elevate the accuracy of knowledge together! 💉💬